Kamerstuk
| Datum publicatie | Organisatie | Vergaderjaar | Dossier- en ondernummer |
|---|---|---|---|
| Tweede Kamer der Staten-Generaal | 2022-2023 | 29477 nr. 838 |
U bent nu hier: Publicaties Officiële publicaties
Zoals vergunningen, bouwplannen en lokale regelgeving.
Adressen en contactpersonen van overheidsorganisaties.

Aan de Voorzitter van de Tweede Kamer der Staten-Generaal
Den Haag, 16 juni 2023
Er zijn veel aandoeningen die pakweg 30 jaar geleden ongeneeslijk waren, maar waarbij het nu mogelijk is deze te behandelen of te genezen. En veel ziekten die we vandaag niet kunnen genezen, kunnen we dankzij onderzoek en nieuwe technieken hopelijk in de nabije toekomst wel genezen of behandelen. Ik hecht aan deze innovatie want met die vooruitgang is voor patiënten steeds vaker een effectieve behandeling beschikbaar.
De uitgaven aan dure geneesmiddelen, die voornamelijk in het ziekenhuis worden gegeven, zijn gestegen van € 1,9 miljard in 2016 naar € 2,6 miljard in 2021.1 De verwachting is dat de uitgavengroei aan deze dure intramurale geneesmiddelen ook na dit jaar doorzet met jaarlijks gemiddeld 5% tot 7%.2 Deze forse uitgavenstijging vindt plaats ondanks de inspanningen die worden geleverd om dure intramurale geneesmiddelen weloverwogen te laten instromen in het basispakket, door middel van de zogenaamde sluisprocedure. Omdat het financieel kader waaruit dure intramurale geneesmiddelen worden bekostigd de komende jaren aanzienlijk minder hard stijgt dan de uitgaven aan intramurale geneesmiddelen, verdringt dit andere vormen van zorg en zet daarmee de solidariteit onder druk.3 Daarbij komt dat bij deze nieuwe dure geneesmiddelen er onzekerheid over de effectiviteit kan zijn. Ik zie dat er in de afgelopen jaren steeds vaker geneesmiddelen geregistreerd worden met een beperkte meerwaarde. Dat vraagt van alle partijen dat zij keuzes maken om de uitgaven te beheersen zodat innovatieve geneesmiddelen beschikbaar en betaalbaar blijven voor patiënten. Het uitgangspunt hiervoor is dat patiënten passende zorg ontvangen.4 Ik doe dat ook in het kader van de afspraken die ik heb gemaakt over dure geneesmiddelen in het Integraal Zorgakkoord (IZA), in lijn met mijn ambitie om de toets op het basispakket te verbeteren en verbreden.5
Hoewel het dure-geneesmiddelenbeleid vele facetten kent, beperk ik mij in deze brief tot de uitdagingen en oplossingsrichting bij de toelating van dure geneesmiddelen tot het basispakket. Ik schets u daartoe: 1) de uitdagingen bij nieuwe dure geneesmiddelen; 2) mijn huidige inzet om de instroom van dure intramurale geneesmiddelen beheersbaar te houden; 3) hoe ik de instroom van deze geneesmiddelen toekomstbestendig wil maken; 4) een aantal randvoorwaarden die bij het pakketbeheer van dure geneesmiddelen noodzakelijk zijn.
Rapport «Van grip naar greep»
Parallel aan deze Kamerbrief bied ik u het rapport «Van grip naar greep, de ex-post evaluatie van de geneesmiddelenvisie» van onderzoeksbureau SiRM aan, inclusief mijn beleidsreactie daarop (Kamerstuk 29 477, nr. 839). Met die evaluatie is gereflecteerd op het beleid en de maatregelen op het gebied van dure geneesmiddelen sinds 2016. De voorstellen in deze brief heb ik geschreven in het licht van de aanbevelingen uit de evaluatie.
Ontwikkelingen en uitdagingen rondom de markttoelating
Ik signaleer dat er bij markttoelating door het Europees Geneesmiddelenbureau (EMA) regelmatig onzekerheid bestaat over de effectiviteit van het betreffende geneesmiddel. Het aantal geneesmiddelen dat de afgelopen jaren door het EMA is toegelaten op basis van een voorwaardelijke handelsvergunning of een handelsvergunning onder uitzonderlijke voorwaarden loopt stapsgewijs op. In 2016 waren dat er 9, in 2022 is dat opgelopen tot 14.6 Dat zijn geneesmiddelen die zijn toegelaten op basis van beperktere bewijslast. Dat betekent dat het EMA bij de registratie accepteert dat er onzekerheid bestaat over de precieze effectiviteit van deze geneesmiddelen. Firma’s moeten na markttoelating de werkzaamheid en/of bijwerkingen van deze geneesmiddelen verder karakteriseren. Deze onzekerheid op het moment van markttoelating werkt door in het bepalen van de relatieve effectiviteit (zie toelichting in tekstbox 1).
Ook zie ik dat firma’s hoge prijzen vragen voor deze nieuw toegelaten geneesmiddelen. Het ontwikkelen van een nieuw molecuul tot een werkzaam geneesmiddel is een ingewikkeld proces dat financieel risico’s met zich meebrengt. Daarom steun ik in principe het systeem waarin firma’s de mogelijkheid hebben om de kosten voor onderzoek, ontwikkeling en risicovolle investeringen terug te verdienen door middel van patenten en aanvullende beschermingsmechanismen. Ik ben echter kritisch over de duur van deze aanvullende beschermingsmechanismen. Steeds vaker zie ik een disbalans tussen de gevraagde prijs voor een nieuw geneesmiddel en de zekerheid dat het geneesmiddel ook doet wat de firma belooft. Dit heeft ertoe geleid dat ik de afgelopen maanden helaas enkele keren genoodzaakt was om te besluiten een geneesmiddel niet op te nemen in het basispakket van de zorgverzekering, omdat de firma’s niet bereid waren om tot een kosteneffectieve en daarmee maatschappelijk aanvaardbare prijs te komen.7
Tekstbox 1. Marktoelating en pakkettoelating nader toegelicht.
Markttoelating en pakkettoelating van sluisgeneesmiddelen nader toegelicht
In de toelating via de sluis van intramurale geneesmiddelen tot het basispakket, wordt vaak verwezen naar twee organisaties:
Het Europees Geneesmiddelenbureau (EMA) en Zorginstituut Nederland (Zorginstituut). Het EMA is verantwoordelijk voor de toelating van geneesmiddelen tot de Europese markt en bepaalt of een geneesmiddel van goede kwaliteit is, of het werkzaam is (treedt er een effect op), en of het veilig is. Het EMA beoordeelt de balans tussen de effecten/werkzaamheid en de bijwerkingen en risico’s van medicijnen. Vaak baseert het EMA zich op studies waarin bij patiënten de beoogde gezondheidseffecten onder optimale onderzoeksomstandigheden worden bereikt. Na het verlenen van een handelsvergunning voor de Europese markt, heeft elke lidstaat een eigen procedure om te bepalen of het geneesmiddel ook wordt vergoed voor patiënten.
Als een duur geneesmiddel onderdeel is van de pakketsluis, dan toetst het Zorginstituut primair of het geneesmiddel voldoet aan «de stand van wetenschap en praktijk». Hierbij wordt gekeken naar de relatieve effectiviteit, waarin beoordeeld wordt of de nieuwe interventie in vergelijking met de huidige standaardbehandeling even goed of beter werkt. Als er gegevens beschikbaar zijn, baseert het Zorginstituut zich daarbij op gegevens of de effecten bij patiënten in de dagelijkse klinische praktijk worden bereikt. Daarnaast wordt getoetst op de kosteneffectiviteit, waarbij gewogen wordt of de extra kosten van een nieuw geneesmiddel in verhouding staan tot de relatieve effectiviteit. De beslissing of een sluisgeneesmiddel uiteindelijk wordt opgenomen in het basispakket, al dan niet onder voorwaarden, is daarna aan de Minister. Om tot opname in het basispakket te komen, kunnen bijvoorbeeld prijsonderhandelingen worden gevoerd om de kosteneffectiviteit te verbeteren.
Ontwikkelingen en uitdagingen rondom de pakkettoelating
Bij de pakkettoelating spelen andersoortige ontwikkelingen en uitdagingen. Voordat dure geneesmiddelen worden vergoed vanuit het basispakket, kunnen deze onderdeel zijn van de zogenaamde sluisprocedure. Deze procedure wordt in de volgende paragraaf nader toegelicht. In de sluisprocedure adviseert het Zorginstituut over de randvoorwaarden waaronder deze geneesmiddelen beheerst kunnen instromen in het basispakket. Maar deze beheerste instroom van dure geneesmiddelen is niet afdoende om grip te houden op de stijgende uitgaven aan dure geneesmiddelen.
Binnen het Integraal Zorgakkoord (IZA) is afgesproken dat in 2023 en 2024 een jaarlijkse uitgavengroei van het kader Medisch Specialistische Zorg van maximaal 7% kan worden opgevangen, en dat in 2025 en 2026 de jaarlijkse uitgavengroei maximaal 5% is. Alle partijen, en dus ook ik, zijn gezamenlijk verantwoordelijk om de uitgaven aan dure geneesmiddelen te beheersen. Als de uitgavengroei van 5% tot 7% wordt overschreden, en ook het totale uitgavenkader voor medisch specialistische zorg wordt overschreden, nemen zowel het Ministerie van VWS als zorgpartijen de helft van de extra uitgaven voor hun rekening. Om te voorkomen dat die extra uitgaven gedaan gaan worden, is in het IZA afgesproken dat extra maatregelen nodig zijn voor de beheerste instroom van dure geneesmiddelen. We hebben daarbij oog voor de kwaliteit, toegankelijkheid en betaalbaarheid van nieuwe dure geneesmiddelen. Het huidige systeem van pakkettoelating is onvoldoende toereikend om deze uitgavenstijging te beheersen en tegemoet te komen aan de uitdagingen in de geneesmiddelenmarkt, zoals het toenemend aantal geneesmiddelen dat voorwaardelijk tot de markt is toegelaten.
Voordat ik inga op de hoofdlijnen om de instroom van dure intramurale geneesmiddelen toekomstbestendig te maken, wil ik een aantal zaken noemen waar nu al op wordt ingezet. Dat zijn op de eerste plaats de eigenstandige inspanningen van het Zorginstituut, en activiteiten van het Zorginstituut, de ACM en de NZa gezamenlijk. Daarnaast licht ik uit welke maatregelen ik zelf al heb ondernomen, zowel in de nationale als internationale context.
Verbeteren en verbreden toets basispakket
Uw Kamer heb ik reeds geïnformeerd over mijn voornemen tot het verbeteren en het verbreden van de toets op het basispakket. Onder verbeteren verstaan we de verdere borging van de huidige criteria effectiviteit, kosteneffectiviteit, noodzakelijkheid en uitvoerbaarheid. Daarbij wordt er gekeken naar het verankeren van aanvullende voorwaarden voor gepast gebruik en onderzoek. Ook duurzaamheid en personeelsinzet moeten een rol spelen bij de weging of zorg vanuit het basispakket vergoed moet worden. Uitgangspunt van verbreden is dat zorg uit alle sectoren, en dus ook geneesmiddelen, vaker (cyclisch) getoetst gaat worden aan de pakketcriteria. De contouren hiervan zijn zichtbaar in enkele recente publicaties van het Zorginstituut. Zo heeft het Zorginstituut haar visie op het pakketbeheer gepubliceerd8 en de beoordeling van «de stand van wetenschap en praktijk» (hierna: SWP) geactualiseerd.9 Daarin is ook een module opgenomen voor de beoordeling van tumoragnostische geneesmiddelen en andere oncologische geneesmiddelen die uitsluitend met een enkelarmige studie zijn onderzocht. Bij tumoragnostische geneesmiddelen is niet de tumorlocatie leidend, maar de moleculaire kenmerken van de tumor.
Het Zorginstituut biedt hiermee een handreiking aan partijen in de beoordeling van deze typen geneesmiddelen. Ook heeft het Zorginstituut informatiemateriaal ontwikkeld bedoeld om de toepassing van de beoordeling van de SWP door veldpartijen te faciliteren.
Beleidsadvies maatschappelijk aanvaardbare prijzen
Daarnaast wordt door de Nederlandse Zorgautoriteit, de Autoriteit Consument en Markt en het Zorginstituut een beleidsadvies opgesteld over een Nederlands kader voor maatschappelijk aanvaardbare prijzen voor dure geneesmiddelen.10 Dit advies is een uitvloeisel van de motie van het lid Kuiken.11 Na oplevering van het advies zal ik besluiten over de wijze waarop dit kader kan bijdragen aan de betaalbaarheid van geneesmiddelen en de solidariteit in het zorgstelsel.
Nederlandse inzet in internationale gremia
Ook in internationale gremia is Nederland actief. Zo stimuleert Nederland dat geneesmiddelen worden ontwikkeld die voorzien in een «unmet medical need». Dit betekent dat we willen dat de geneesmiddelenmarkt meer «vraaggericht» wordt, zodat er vooral geneesmiddelen worden ontwikkeld voor ziekten waarvoor nog geen afdoende (effectieve) behandeling bestaat. Daarnaast werken het Ministerie van VWS en het Zorginstituut actief samen met andere Europese landen aan het vraagstuk dure geneesmiddelen. Er wordt bijvoorbeeld gewerkt aan gezamenlijke beoordelingen van de effectiviteit van geneesmiddelen, op basis van health technology assessments. Op deze wijze kan het proces van toelating tot het verzekerde pakket versnellen. Ook wordt in het Beneluxa-verband op vrijwillige basis samengewerkt aan prijsonderhandelingen.
Aanpassing sluiscriteria
Ondanks het internationale karakter van de geneesmiddelenontwikkeling zijn we in Nederland primair aan zet om zelf keuzes te maken welke geneesmiddelen we vergoeden. Het belangrijkste instrument daartoe is de beoordeling van de effectiviteit, kosteneffectiviteit, noodzakelijkheid en uitvoerbaarheid van nieuwe geneesmiddelen. Deze beoordelingen bieden de mogelijkheid om financiële arrangementen te sluiten met firma’s van geneesmiddelen. In 2018 is hiervoor een specifiek wettelijk kader geïntroduceerd; de zogenoemde sluis. Daarmee worden dure intramurale geneesmiddelen uitgesloten van de open instroom in het basispakket. Recentelijk heb ik bekendgemaakt dat de criteria voor toepassing van de sluis per 1 juli 2023 worden aangescherpt.12 Vanaf die datum kan een geneesmiddel in twee gevallen in aanmerking komen voor de sluisprocedure:
– Wanneer het verwachte macrokostenbeslag van het geneesmiddel voor één of meerdere nieuwe indicaties samen jaarlijks € 20 miljoen of meer bedraagt. In dat geval worden alle nieuwe en toekomstige indicaties in de sluis geplaatst.
– Wanneer de verwachte kosten van het geneesmiddel voor een nieuwe indicatie jaarlijks € 50.000 of meer per patiënt zijn én het verwachte macrokostenbeslag jaarlijks € 10 miljoen of meer bedraagt. In dat geval wordt het geneesmiddel alleen voor de nieuwe indicatie in de sluis geplaatst.
Van deze geneesmiddelen acht ik het niet verantwoord om ze zonder meer in te laten stromen in het basispakket. Gedurende de toetsing door het Zorginstituut maken sluisgeneesmiddelen geen deel uit van het basispakket. Pas als deze geneesmiddelen aan de pakketcriteria voldoen, kan opname in het basispakket plaatsvinden. Een financieel arrangement kan eraan bijdragen dat een geneesmiddel kosteneffectief wordt ingezet en daarmee pakketwaardig is. In de afgelopen jaren zijn goede financiële resultaten behaald met de sluisprocedure. In 2020 bedroeg de uitgavenverlaging door de toepassing van de sluis op dure intramurale geneesmiddelen € 383 miljoen.13
Tekstbox 2. Toegang tot sluisgeneesmiddelen nader toegelicht.
Hoe kan een patiënt in Nederland toegang krijgen tot een nieuw duur geneesmiddel tijdens de sluisprocedure?
Als een geneesmiddel aan de criteria voor de sluisprocedure voldoet (zie toelichting hierboven), dan wordt het in de sluis geplaatst. Daarmee is het voorlopig uitgezonderd van het basispakket. Na advisering door het Zorginstituut of een sluisgeneesmiddel in het basispakket opgenomen moet worden, volgt vaak nog een prijsonderhandeling met de firma van het geneesmiddel. Gedurende de «sluisperiode» wordt het sluisgeneesmiddel niet vergoed vanuit de zorgverzekering. Gelukkig stellen de meeste firma’s hun geneesmiddel op eigen kosten beschikbaar voor patiënten die niet kunnen wachten. Om ervoor te zorgen dat firma’s dat ook in de toekomst blijven doen, is het Ministerie van VWS bereid om deze kosten – onder voorwaarden – mee te wegen bij het afsluiten van een financieel arrangement.
Dure extramurale geneesmiddelen
Hoewel ik mij met deze Kamerbrief primair richt op de dure intramurale geneesmiddelen, informeer ik u dat ook voorafgaand aan de opname van dure extramurale geneesmiddelen het Zorginstituut meer adviezen gaat uitbrengen die kunnen leiden tot prijsonderhandeling. Dure extramurale geneesmiddelen worden pas vergoed als ze zijn opgenomen in het geneesmiddelenvergoedingssysteem (GVS). Voordat een geneesmiddel kan worden opgenomen in het GVS, adviseert het Zorginstituut over pakketopname.
Een recente wijziging in het beleid is dat het Zorginstituut vaker een beoordeling gaat uitvoeren naar de kosteneffectiviteit van extramurale geneesmiddelen. Daarvoor is de grens voor de zogenaamde farmaco-economische vrijstelling aangepast. Het Zorginstituut beziet daarvoor het macrokostenbeslag in plaats van de budgetimpact.14 Bovendien wordt de grenswaarde voor het uitvoeren van een farmaco-economische beoordeling verlaagd naar:
– Als het macrokostenbeslag € 10 miljoen of meer per jaar bedraagt, of;
– Het macrokostenbeslag tussen de € 1 miljoen en € 10 miljoen per jaar bedraagt en de kosten per patiënt per jaar hoger zijn dan € 50.000.
Als uit de kosteneffectiviteitsbeoordeling volgt dat een extramuraal geneesmiddel niet kosteneffectief is, dan wordt een geneesmiddel niet opgenomen in het GVS. Primair is mijn inzet dat er openbare prijsverlagingen plaatsvinden zodat deze extramurale geneesmiddelen pakketwaardig worden. In specifieke gevallen is dat niet mogelijk. Dan kan het zijn dat een financieel arrangement een voorwaarde is om op te worden genomen in het GVS.
Tekstbox 3. Toegang tot geneesmiddelen nadat ze zijn toegelaten tot het basispakket nader toegelicht.
Hoe kan een patiënt in Nederland toegang krijgen tot een nieuw duur geneesmiddel nadat het is toegelaten tot het basispakket?
In de periode na de sluisprocedure ligt de nadruk op de inzet in de praktijk en het «gepast gebruik» van geneesmiddelen. Hiervoor zijn per ziektegebied zogenaamde beroepsverenigingen waarin artsen met elkaar beslissen of zij bijvoorbeeld een nieuw geneesmiddel in Nederland gaan toepassen, welke patiënten daarvoor in aanmerking komen, en welke criteria ze gebruiken om ook weer te stoppen met een behandeling. De meest bekende toetsingscriteria zijn die van de vereniging van oncologen (de NVMO); deze zijn openbaar, en zijn recent aangescherpt. Met openbare toetsingscriteria worden alle nieuwe kankergeneesmiddelen op dezelfde wijze gewogen, en wordt gewaarborgd dat elke arts een patiënt op dezelfde manier behandelt en dat dat gebeurt op basis van de meest recente wetenschappelijke inzichten.
Van sommige nieuwe geneesmiddelen, bijvoorbeeld van gentherapieën, is het relatief makkelijk te bepalen wie voor een nieuw geneesmiddel in aanmerking komt. Zo kan gekeken worden naar de genetische afwijking, en is vooral van belang of de gezondheidsconditie van een patiënt goed genoeg is om eventuele bijwerkingen van een behandeling te kunnen dragen. In andere gevallen, bijvoorbeeld bij nieuwe geneesmiddelen voor de behandeling van kanker, kan dat minder duidelijk zijn. Vaak zijn er al behandelingen beschikbaar, en moet bepaald worden wanneer patiënten behandeld worden met een geneesmiddel; vervangt een nieuw geneesmiddel de huidige behandeling, of volgt het nieuwe geneesmiddel op de bestaande behandeling? Vooral als een nieuw geneesmiddel wel levensverlengend is, maar geen kans op genezing geeft, vinden artsen het extra belangrijk om te weten wat de (kans op) bijwerkingen van een nieuw geneesmiddel zijn.
Lang niet alle middelen maken onderdeel uit van de sluisprocedure. Dat komt omdat de kosten voor deze geneesmiddelen onder de grenswaarden van de sluis kunnen liggen, bijvoorbeeld omdat er sprake is van een hoge prijs en een zeer gering aantal patiënten. Deze geneesmiddelen worden op dit moment primair beoordeeld door zorgverzekeraars. Zij geven aan veel moeite te hebben met het vaststellen van de effectiviteit van deze middelen. Daar komt bij dat het voor hen lastig is om bij dergelijke geneesmiddelen tot prijsafspraken te komen, omdat zij afhankelijk zijn van de bereidheid van firma’s om deze afspraken te maken. Ik signaleer daarom het risico dat deze geneesmiddelen niet (kosten)effectief in het basispakket instromen. Dat betekent dat ik niet altijd goed weet wat de meerwaarde van deze geneesmiddelen is voor patiënt en premiebetaler.
Dat onderstreept wat mij betreft de noodzaak om aanvullende maatregelen te treffen bij de instroom van dure geneesmiddelen. Ik wil, voordat een duur geneesmiddel deel uitmaakt van het basispakket, bezien wat te doen als deze dure geneesmiddelen bedoeld zijn voor een gering aantal patiënten, een (relatief) hoge prijs hebben, er onzekerheid is over effectiviteit, en/of er een vraag is rondom gepast gebruik. Ik doe dit als onderdeel van het traject van het verbeteren en het verbreden van de toets op het basispakket. De aanvullende maatregelen borduren voort op bestaand instrumentarium, maar ik wil deze beter verankeren en structureel inzetten. Ik onderscheid daarbij drie fasen die bijdragen aan een toekomstbestendig intramuraal stelsel voor dure geneesmiddelen en daarmee invulling geven aan de afspraken uit het IZA. De eerste twee fasen richten zich op het identificeren van risico’s.
In fase 1 worden, rondom de markttoelating, in korte tijd risico’s gekwantificeerd door het Horizonscanteam van het Zorginstituut. In fase 2 worden die risico’s nader geduid in een rapid review. De derde fase richt zich op het beheersen van de geïdentificeerde risico’s voordat een geneesmiddel tot het verzekerde pakket wordt toegelaten. De eerste twee fasen zijn een werkwijze die voortvloeit uit het verbreden van de toets op het basispakket; de derde fase is een uitwerking van het verbeteren van de toets op het basispakket. Ik zie alle fasen in dit proces als een gezamenlijke verantwoordelijkheid van de partijen verenigd in het Landelijk Overleg Dure Geneesmiddelen. In figuur 1 schets ik u op hoofdlijnen hoe ik dit proces voor mij zie. Ik licht dit onder de figuur nader toe.
Figuur 1. Proces op hoofdlijnen van beheerste instroom van geneesmiddelen.
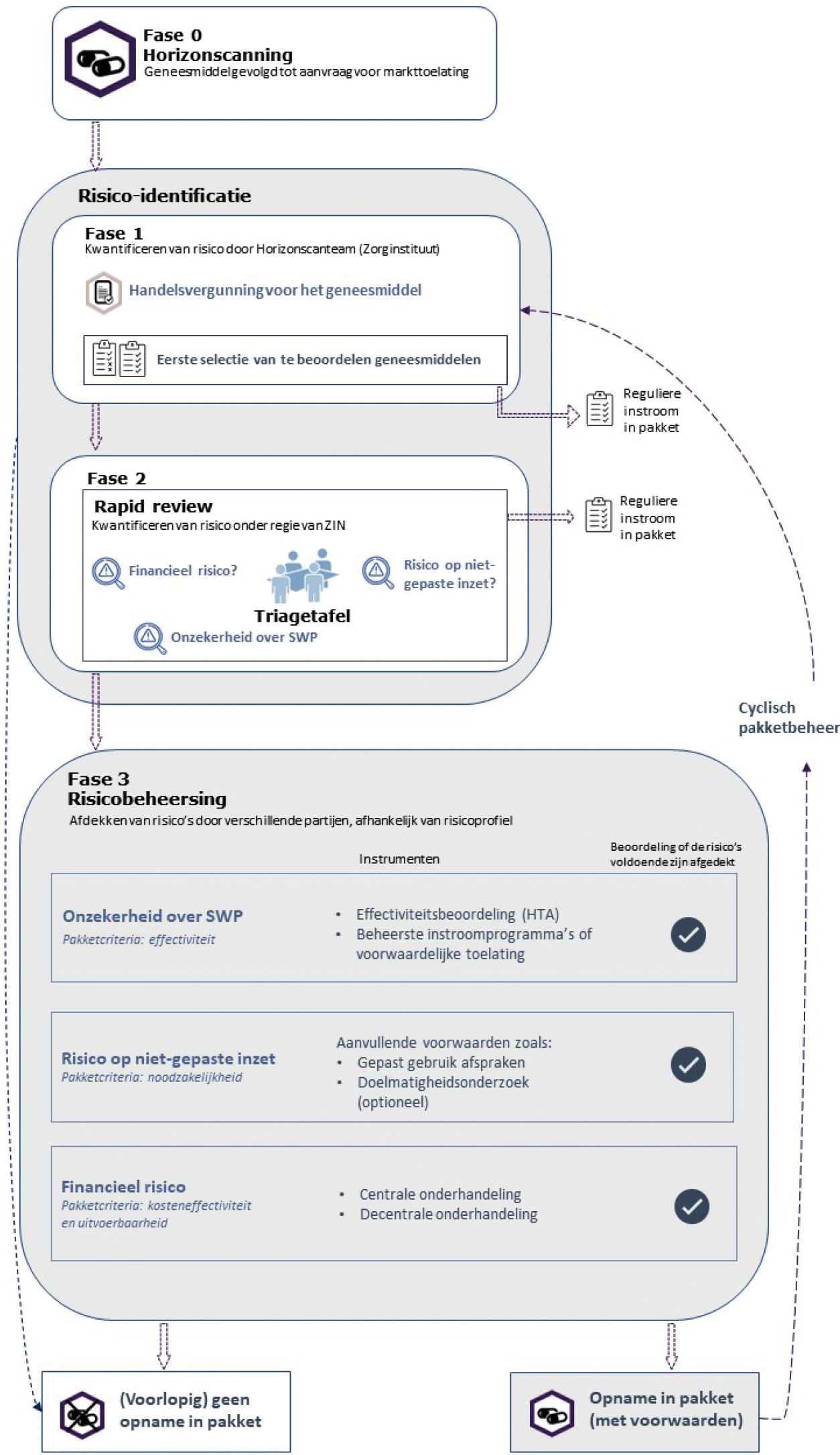
Fase 0 – Horizonscanning
Met de bestaande Horizonscan dure geneesmiddelen bestaat er in Nederland een solide basis om te monitoren welke innovatieve geneesmiddelen de komende jaren op de Nederlandse markt komen. In figuur 1 wordt de Horizonscan benoemd onder «fase 0». Mijn ambitie is om deze fase, met kleine aanpassingen, voort te zetten in de beheerste instroom.
Fase 1 – Risico-identificatie
Fase 1 zie ik als een aanpassing van de bestaande procedure waarin geneesmiddelen in de sluis voor dure geneesmiddelen worden geplaatst. Deze stap heet in het nieuwe systeem «risico-identificatie» en vindt plaats rondom het moment van markttoelating van een nieuw geneesmiddel. In principe volgt voor alle nieuwe innovatieve geneesmiddelen een eerste selectie van de aanwezigheid van risico’s op basis van feitelijke informatie waarin deze geneesmiddelen worden beoordeeld. Daarvoor worden nieuwe gestandaardiseerde en eenduidige criteria gebruikt. Op die manier kan snel bepaald worden of een nieuw geneesmiddel direct kan worden opgenomen in het verzekerde pakket, of dat risico’s nader bekeken moeten worden. Dat gebeurt met het doorlopen van het proces van de beheerste instroom, voordat het geneesmiddel kan worden opgenomen in het basispakket. In het geval er geen of beperkte risico’s zijn, dan volgt de reguliere instroom in het basispakket. Daarin zijn zorgverzekeraars aan zet om zo snel als mogelijk te beoordelen of patiënten in aanmerking komen voor vergoeding van het geneesmiddel.
Fase 2 – Rapid review en triagetafel
Fase 2 is een nieuwe stap in de beheerste instroom. In het geval fase 1 van de risico-identificatie aanleiding geeft om een geneesmiddel niet direct toe te laten tot het basispakket, gaat het geneesmiddel door naar fase 2. In die fase worden de risico’s nader in kaart gebracht met behulp van een rapid review. Ik voorzie dat deze rol onder regie bij het Zorginstituut wordt belegd, maar waarbij de input van partijen zoals de beroepsgroep en zorgverzekeraars zeer belangrijk is. Zij kunnen meer informatie aanleveren over de marktstatus, het verwachte macrokostenbeslag en het aantal patiënten dat in Nederland in aanmerking komt voor het geneesmiddel. In deze fase wordt ook literatuur en geldende behandelrichtlijnen betrokken, waarbij ik voorzie dat gekeken gaat worden naar de indicatie, aangetoonde effectiviteit, epidemiologische gegevens en het bestaan van vergelijkbare geneesmiddelen (nu of toekomstig). Voor de rapid review is verder van belang dat firma’s gegevens aanleveren over onder meer de effectiviteit en de prijs van het geneesmiddel.
Als de rapid review is opgesteld, vindt in fase 2 een bespreking plaats aan de triagetafel. Het doel van de triagetafel is om te beslissen welke vervolgstappen in fase 3 van de beheerste instroom moeten worden genomen. Sowieso kan in deze fase alsnog besloten worden dat de risico’s beheersbaar zijn, en dat het geneesmiddel onderdeel wordt van de reguliere instroom in het basispakket. Indien dat niet het geval is, voorzie ik dat partijen in gezamenlijkheid aan de zogenaamde triagetafel de geïdentificeerde risico’s nader duiden. Het gaat hierbij om drie typen risico’s:
– Onzekerheid over «de stand van wetenschap en praktijk». De triagetafel kan constateren dat een risico bestaat dat een geneesmiddel niet, of niet voor alle patiënten, effectief is. In dat geval zal geadviseerd worden dat het Zorginstituut een uitgebreide effectiviteitsbeoordeling gaat uitvoeren, zoals dat nu ook het geval is bij alle sluisgeneesmiddelen.
Een andere uitkomst van de triagetafel kan zijn dat zij een risico signaleren, al dan niet boven op een effectiviteitsbeoordeling, en er ingezet moet worden op een programma voor beheerste instroom of voorwaardelijke toelating.
– Risico’s op niet-gepaste inzet. De triagetafel kan constateren dat risico’s bestaan met betrekking tot de gepaste inzet van een geneesmiddel. Dat kan bijvoorbeeld zijn omdat de plaats van het geneesmiddel in een behandelrichtlijn nader moet worden uitgezocht. Ook kan het zijn dat afspraken moeten worden gemaakt over de concentratie van zorg. Dat draagt bij aan effectievere zorg, bijvoorbeeld als het gaat om een duur weesgeneesmiddel. Anderzijds kan het zijn dat geconstateerd wordt dat, in het belang van de patiënt, het belangrijk is om start-stopcriteria of een (hybride) doseringsadvies te formuleren voordat een geneesmiddel wordt opgenomen in het basispakket. Ook kan het zijn dat – bij de opname van een geneesmiddel in het basispakket – direct gestart moet worden met een aanvullend doelmatigheidsonderzoek omdat er veel potentie is om de kwaliteit of effectiviteit van een geneesmiddel te verhogen.
– Financiële risico’s. De triagetafel kan constateren dat er risico’s zijn dat een geneesmiddel niet kosteneffectief is, en dat de kosteneffectiviteit nader onderzocht moet worden. Ook kan het zijn dat de triagetafel vaststelt dat er een risico is dat de budgetimpact door opname van een geneesmiddel in het verzekerde pakket de uitvoerbaarheid van de zorgverzekering in het geding brengt.
Het is van belang om op te merken dat in fase 2 wordt vastgesteld dat een geneesmiddel één of meerdere van de genoemde risico’s heeft en dat in fase 3 alle risico’s afgedekt moeten zijn voordat het geneesmiddel opgenomen kan worden in het basispakket.
Voor zowel fase 1 en fase 2 van de risico-identificatie geldt dat de samenwerking van alle betrokken partijen essentieel is, en ik daarbij verwacht dat ieder met zijn eigen expertise en verantwoordelijkheid kan aansluiten. Ik voorzie op dit moment dat het Zorginstituut de voorzitter van dit proces wordt. De andere betrokken partijen van dit proces zijn behandelaren (artsen en apothekers), ziekenhuizen, patiënten en zorgverzekeraars. Ik wil de exacte rollen en verantwoordelijkheden van de beroepsgroepen, ziekenhuizen, patiënten, zorgverzekeraars, het Zorginstituut en het Ministerie van VWS bij de verschillende stappen in het proces nader met hen uitwerken. Het ligt voor de hand dat daarbij wordt doorgewerkt op elementen uit de huidige systematiek van de sluis voor intramurale geneesmiddelen. Ik denk hierbij aan de werkgroepen van de Horizonscan waar behandelaren (artsen en apothekers), vertegenwoordigers van zorgverzekeraars en patiënten deel van uitmaken. Ook zie ik een belangrijke rol voor de geneesmiddelencommissies die door de Federatie Medisch Specialisten bij de wetenschappelijke verenigingen worden ingericht. Tot slot ben ik voornemens om, daar waar nodig, ook vertegenwoordigers van de farmaceutische firma’s te betrekken bij de uitwerking.
Fase 3 is een grotendeels een nieuwe stap in het pakketbeheer van dure geneesmiddelen. Wanneer de risico’s zijn geïdentificeerd, start fase 3, het proces van risicobeheersing. Dit betekent dat, afhankelijk van welke risico’s geïdentificeerd zijn, verschillende stappen worden ondernomen om de risico’s af te dekken.
Onzekerheid over SWP en/of niet-gepaste inzet
Als uit de beoordeling van het Zorginstituut blijkt dat er onzekerheid bestaat over «de stand van wetenschap en praktijk» of eerder is gesignaleerd dat er een risico bestaat op niet-gepaste inzet van een geneesmiddel, dan ligt in beide gevallen het voor de hand dat veldpartijen onderling nadere afspraken maken. Deze afspraken kunnen gaan over het doen van vervolgonderzoek naar de (lange termijn) effectiviteit van het geneesmiddel of naar bijvoorbeeld start- en stopcriteria of de dosering binnen een bepaalde periode. Maar het kan ook gaan over het maken van afspraken over de gepaste inzet van het geneesmiddel (wie krijgt wanneer het middel), zodat het geneesmiddel onder die voorwaarde opgenomen kan worden in het basispakket. Dit is tevens een uitwerking binnen de context van dure geneesmiddelen van de ambitie die ik in het bredere traject van het verbeteren en het verbreden van de toets op het basispakket heb uitgesproken. Daarin wil ik het mogelijk en juridisch afdwingbaar maken om aanvullende voorwaarden te stellen aan vergoeding vanuit het basispakket.
Financiële risico’s
Het afdekken van de financiële risico’s betekent dat de uitgaven aan het betreffende geneesmiddel door middel van financiële afspraken beheerst moeten worden voordat opname in het basispakket kan plaatsvinden. In dit proces wil ik expliciet aandacht besteden aan zowel centrale onderhandelingen door het BBuro Financiële Arrangementen Geneesmiddelen van het Ministerie van VWS als decentrale prijsonderhandelingen door zorgverzekeraars of ziekenhuizen. De keuze daarvoor hangt af van het betreffende geneesmiddel en bijvoorbeeld het bestaan van concurrentie. Voor het goed afdekken van deze risico’s kan het in gevallen nodig zijn om in aanvulling op de rapid review een volledige pakketbeoordeling door het Zorginstituut uit te voeren. In andere gevallen kan het afdoende zijn als zorgverzekeraars deze beoordeling uitvoeren, al dan niet in samenwerking met beroepsgroepen en patiëntvertegenwoordigers.
Het besluit of een duur geneesmiddel wordt opgenomen in het basispakket, ligt bij mij als Minister van VWS. Voordat ik een dergelijk besluit neem, wil ik laten beoordelen of de gesignaleerde risico’s in voldoende mate zijn afgedekt. Dat betekent dat gecontroleerd wordt of de eerder geïdentificeerde risico’s van de triagetafel voldoende zijn afgedekt voor een verantwoorde opname in het verzekerde pakket. Voor die geneesmiddelen waarbij via een centraal financieel arrangement de financiële risico’s worden afgedekt, geldt dat deze verificatiestap binnen het Ministerie van VWS plaatsvindt, waarna ik als Minister besluit tot opname in het basispakket.
Cyclisch pakketbeheer
Om het gehele proces af te sluiten, wil ik in de nadere uitwerking afspraken maken over het structureel inbedden van cyclisch pakketbeheer. Dit waarborgt bijvoorbeeld dat afspraken gemonitord kunnen worden, maar ook dat geneesmiddelen opnieuw beoordeeld kunnen worden om te bepalen of de risico’s zijn weggenomen en of niet nieuwe risico’s zijn ontstaan door veranderingen in de markt of in het behandellandschap.
Het kan hierbij bijvoorbeeld gaan om herbeoordelingen van geneesmiddelen waarbij onder de aanvullende voorwaarden informatie is verzameld over de effectiviteit van dat geneesmiddel op de lange termijn. Voor sommige middelen kan hier farmacovigilantie-data voor worden gebruikt. Farmacovigilantie (geneesmiddelenbewaking) betreft het bewaken van de veiligheid, werkzaamheid en risico’s nadat een geneesmiddel is geregistreerd. Het aanleveren van deze data is een verplichting van iedere firma. Waar mogelijk kan dit worden gecombineerd met de praktijkervaring en data (bijvoorbeeld nieuwe informatie over biomarkers) van de beroepsgroep(en).
Naast het hierboven beschreven proces, ben ik voornemens enkele randvoorwaarden voor de beheerste instroom van dure geneesmiddelen op orde te brengen zodat de beheerste instroom beter gaat verlopen en vaker tot gewenste uitkomsten leidt.
Op de eerste plaats krijg ik steeds meer signalen en voorbeelden dat de afbakening voor problemen zorgt. Het gaat dan om de vraag of een geneesmiddel extramuraal of intramuraal bekostigd moet worden. De huidige afbakeningscriteria kunnen het doelmatig voorschrijven in de weg staan. Hiervan is bijvoorbeeld sprake wanneer een nieuwe toedieningsvorm van een geneesmiddel extramuraal wordt bekostigd, terwijl het oorspronkelijke middel intramuraal wordt bekostigd. Door deze verschillende bekostigingskaders vermindert de prikkel tot het doelmatig gebruik van het geneesmiddel. Ik ben voornemens deze afbakening nader te bekijken, zodat de afbakening doelmatigheid en concurrentie niet in de weg zit. Ik zal daarbij ook (niet-oncologische) weesgeneesmiddelen bekijken.
Ten tweede is het voor partijen niet altijd duidelijk welke routes tot het basispakket van de zorgverzekering er voor geneesmiddelen bestaan en welke geneesmiddelen voor welke indicatie vergoed worden vanuit het basispakket. Dit gebrek aan duidelijkheid en transparantie is ongewenst, zoals ook in de motie van het Kamerlid Den Haan wordt gesteld.15 Ik verwacht in het najaar te komen met een dashboard voor sluisgeneesmiddelen dat inzichtelijk maakt waar in het vergoedingsproces een geneesmiddel zich bevindt en hoe lang de doorlooptijd is. Ook ben ik met de zorgverzekeraars in gesprek om hun beoordelingsproces transparanter te maken. Zij hebben mij aangegeven dit ook van groot belang te vinden. Daarnaast wil ik ter bevordering van de transparantie ook dat op centraal niveau inzichtelijk is wat de vergoedingsstatus is van intramurale add-on geneesmiddelen. Dat kan met een centrale vergoedingslijst, of door in ieder geval een centrale database te maken met de vergoedingsstatus van add-on geneesmiddelen. Ik ben hierover in gesprek met zorgverzekeraars. Voor extramurale geneesmiddelen is dit overzicht al beschikbaar via medicijnkosten.nl.
Gegeven het bovengenoemde proces zijn in elk geval nog twee aanvullende randvoorwaarden van belang. Allereerst moet de data-infrastructuur op orde zijn, zodat op efficiënte wijze informatie verzameld kan worden over bijvoorbeeld het gepast gebruik van geneesmiddelen of de effectiviteit op de lange termijn. Het belang van een data-infrastructuur in het kader van dure geneesmiddelen is één van de aanbevelingen van de ex-post evaluatie van de geneesmiddelenvisie. Over mijn inzet op het secundair gebruik van gezondheidsgegevens heb ik u reeds in april geïnformeerd.16
Daarnaast is de bijdragen van partijen aan het geschetste proces van groot belang voor het slagen van het proces en het effect op de betaalbaarheid en toegankelijkheid van geneesmiddelen. Daarom is het ook van belang dat partijen voldoende uitgerust zijn om hieraan hun bijdrage te leveren. Het gaat hierbij niet alleen over voldoende capaciteit bij het Zorginstituut, het BBuro Financiële Arrangementen Geneesmiddelen van het Ministerie van VWS en de zorgverzekeraars. Het gaat juist ook om de uitrusting van de andere relevante partijen, zoals de behandelaren en patiënten. Ik ga daarover met de verschillende partijen in gesprek tijdens de uitwerking van de plannen.
Verder heb ik in het vervolg van dit traject ook aandacht voor monitoring en evaluatie van de maatregelen die ik neem en bijvoorbeeld het effect op de uitgaven aan dure geneesmiddelen. Ik zal dit in het vervolgtraject nader uitwerken en neem daarin de geleerde lessen uit de ex-post evaluatie van de geneesmiddelenvisie mee.
Tot slot
Deze brief vormt een eerste belangrijke stap als het gaat om het toekomstbestendig maken van het pakketbeheer voor dure intramurale geneesmiddelen, maar ik realiseer mij dat er nog veel nodig is om deze plannen nader uit te werken en te concretiseren. Daarin is de samenwerking met partijen van het Landelijke Overleg Dure Geneesmiddelen van groot belang. Met hen wil ik verder uitwerken en nader bepalen wat noodzakelijk is om dit plan te realiseren. Ik informeer u in het eerste kwartaal van 2024 over de nadere uitwerking van plan, wat er nodig is om het realiseren en op welke termijn inwerkingtreding kan plaatsvinden.
De Minister van Volksgezondheid, Welzijn en Sport, E.J. Kuipers
Dit betreft de uitgaven aan intramurale geneesmiddelen die meer kosten dan 1.000 euro per patiënt per jaar en die eigenstandig gedeclareerd kunnen worden (add-on geneesmiddelen). Via https://www.gipdatabank.nl/
Zie onder andere https://www.zorginstituutnederland.nl/actueel/nieuws/2018/04/23/zorginstituut-geeft-beeld-van-verdringing-in-de-zorg en NZa. (2022) Monitor medisch-specialistische zorg 2022.
Zie https://www.ema.europa.eu/en/documents/annual-report/2016-annual-report-european-medicines-agency_en.pdf en https://www.ema.europa.eu/en/documents/annual-report/2022-annual-report-european-medicines-agency_en.pdf.
Kamerstuk 29 477, nr. 798. Tot 1 juli 2023 is de grens voor plaatsing in de sluis als de jaarlijkse uitgaven voor één of meer nieuwe indicaties € 40 miljoen of meer bedragen. Die grenswaarde wordt € 20 miljoen na 1 juli 2023.
Het macrokostenbeslag ziet toe op de verwachte bruto uitgaven aan een geneesmiddel. Bij budgetimpact wordt alleen naar de meerkosten gekeken en bijvoorbeeld substitutie met andere geneesmiddelen meegenomen.
Kopieer de link naar uw clipboard
https://zoek.officielebekendmakingen.nl/kst-29477-838.html
De hier aangeboden pdf-bestanden van het Staatsblad, Staatscourant, Tractatenblad, provinciaal blad, gemeenteblad, waterschapsblad en blad gemeenschappelijke regeling vormen de formele bekendmakingen in de zin van de Bekendmakingswet en de Rijkswet goedkeuring en bekendmaking verdragen voor zover ze na 1 juli 2009 zijn uitgegeven. Voor pdf-publicaties van vóór deze datum geldt dat alleen de in papieren vorm uitgegeven bladen formele status hebben; de hier aangeboden elektronische versies daarvan worden bij wijze van service aangeboden.