Aanhangsel van de Handelingen
| Datum publicatie | Organisatie | Vergaderjaar | Nummer | Datum ontvangst |
|---|---|---|---|---|
| Tweede Kamer der Staten-Generaal | 2021-2022 | 923 |
U bent nu hier: Publicaties Officiële publicaties
Zoals vergunningen, bouwplannen en lokale regelgeving.
Adressen en contactpersonen van overheidsorganisaties.

Bent u bekend met het beoordelingsrapport van het Europees Geneesmiddelenbureau (EMA) voor het COVID-19 mRNA-vaccin van Pfizer (verder: EMA-rapport en Pfizer-vaccin), dat deels openbaar gemaakt is en dat de wetenschappelijke evaluatie van de voorwaardelijke handelsvergunning voor dit middel beschrijft?1
Weet u dat het EMA-rapport een beknopte beschrijving geeft van de in de artikelen 4 en 4 bis van Richtlijn 65/65/EEG, in de bijlage van Richtlijn 75/318/EEG en in artikel 2 van Richtlijn 75/319/EEG bedoelde gegevens en bescheiden, waarvan een aanvraag van een vergunning voor een geneesmiddel voor menselijk gebruik vergezeld moet zijn?2, 3, 4
Het rapport van de European Medicines Agency (hierna: EMA) bevat geen verwijzing naar deze richtlijnen, omdat alle drie deze richtlijnen sinds 17 december 2001 niet meer in werking zijn.
Erkent u dat op basis van dit EMA-rapport geconcludeerd kan worden dat bij afgifte van de vergunning niet volledig aan de in vraag 2 genoemde Europese richtlijnen genoemde vereisten werd voldaan, en dat de Committee for Medicinal Products for Human use (CHMP) haar positieve advies voor een voorwaardelijke handelsvergunning voor het Pfizer-vaccin moet hebben uitgebracht op grond van Europese regelgeving die daarin voorziet?
Weet u dat Verordening (EEG) 2309/93 en Richtlijn 91/507/EEG een wettelijke basis bieden om in uitzonderlijke omstandigheden en onder voorwaarden een handelsvergunning af te kunnen geven?5, 6
Nee, gegeven dat deze regelingen niet meer in werking zijn (Verordening (EEG) 2309/93 niet meer sinds 19 november 2005, en Richtlijn 91/507/EEG niet meer sinds 17 december 2001) en daarmee niet langer een wettelijke basis kunnen vormen.
Kunt u bevestigen dat de handelsvergunning voor het Pfizer-vaccin is afgegeven op voorwaardelijke basis vanwege de uitzonderlijke omstandigheid van de pandemische situatie en de urgente noodzaak voor dit product, zoals dit in het EMA-rapport herhaaldelijk gesteld wordt?
Voor het Pfizer-vaccin (ook wel: Comirnaty) geldt dat een voorwaardelijke handelsvergunning is verleend. Nadat de firma aan de voorwaarden heeft voldaan, wordt deze voorwaardelijke handelsvergunning omgezet in een standaard handelsvergunning. Een voorwaardelijke handelsvergunning kan alleen worden verleend om te voorzien in onvervulde medische behoeften voor een geneesmiddel voor de behandeling, preventie of diagnose van een ernstig invaliderende of levensbedreigende ziekte.7 De voorwaardelijke handelsvergunning is afgegeven in het belang van de volksgezondheid, omdat het vaccin een geneesmiddel is dat wordt ingezet voor de voorkoming (profylaxe) van een levensbedreigende ziekte die op geen andere wijze kan worden voorkomen («unmet need») en omdat de voordelen van de onmiddellijke beschikbaarheid van het Pfizer-vaccin groter zijn dan de risico’s van het Pfizer-vaccin.
Weet u dat de op grond van dit EMA-rapport voorwaardelijke handelsvergunning betrekking heeft op het Pfizer-vaccin voor COVID-19, en dat dit middel nog geen officiële wettelijke goedkeuring heeft in Nederland en binnen de EU?
Dit is onjuist: een voorwaardelijke handelsvergunning is gewoon een wettelijke goedkeuring die na publicatie van het besluit door de Europese Commissie direct van kracht is in alle lidstaten van de Europese Unie.
Weet u dat het Pfizer-vaccin voor COVID-19 momenteel nog niet aan de vereisten voldoet om officiële wettelijke goedkeuring te verkrijgen in Nederland en binnen de EU?
Dit is onjuist: zoals ik in mijn antwoord op vraag 5 aangeef, voldoet het Pfizer-vaccin aan de wettelijke vereisten.
Alle COVID-19-vaccins die in Nederland worden toegediend hebben een voorwaardelijke handelsvergunning en voldoen aan de wettelijke eisen voor goedkeuring.
Bent u van mening dat deze voorwaardelijke handelsvergunning voor Nederland moet worden ingetrokken wanneer de in vraag 5 genoemde uitzonderlijke omstandigheid niet meer van toepassing is? Zo nee, waarom niet?
Zoals ik in mijn antwoord op vraag 5 aangeef, is de voorwaardelijke handelsvergunning afgegeven omdat het vaccin een geneesmiddel is dat wordt ingezet voor de voorkoming (profylaxe) van een levensbedreigende ziekte.
De in vraag 5 genoemde uitzonderlijke omstandigheid is dan ook niet de grondslag van deze voorwaardelijke handelsvergunning.
Bent u van mening dat de voorwaardelijke handelsvergunning voor Nederland moet worden ingetrokken indien de van toepassing zijnde Europese regelgeving niet in acht wordt genomen? Zo nee, waarom niet?
In een situatie waarin op basis van beschikbaar gekomen informatie een geneesmiddel mogelijk niet voldoet aan de wettelijke eisen, zal het EMA een onderzoek instellen. Als de fabrikant niet voldoet aan de gestelde voorwaarden, zal de Europese Commissie op advies van beoordelingscomités van het EMA (CHMP, PRAC) de (voorwaardelijke) de handelsvergunning opschorten of andere maatregelen nemen. Dit werkt dan van rechtswege door in Nederland.
Kunt u uitleggen waarom de in de bijlage van richtlijn 91/507/EEG, deel 4, onder G gestelde voorwaarde, dat het middel alleen op medisch voorschrift mag worden verstrekt en in bepaalde gevallen alleen onder streng medisch toezicht door een bevoegd persoon mag worden toegediend, in Nederland niet wordt toegepast bij de toediening van de COVID-vaccins aan de gehele Nederlandse bevolking?8
Richtlijn 91/507/EEG is sinds 17 december 2001 niet meer in werking en dus wordt geen enkel aspect uit deze regelgeving nog toegepast in Nederland.
Weet u dat het EMA-rapport een opsomming geeft van een vijftal uit ernstige bezwaren voortkomende specifieke verplichtingen (SO's) die de indiener van de aanvraag moet opleveren, inclusief de opleverdata van de 29 aan deze SO's verbonden deelproducten (blz. 36–39) en dat er daarnaast 23 aanbevelingen worden gedaan voor de toekomstige kwaliteitsontwikkeling (blz. 39–41)?
Ja, in het EMA-rapport worden op pag. 36–41 een aantal specifieke verplichtingen (specific obligations; SO’s) en aanbevelingen gegeven. Ik kan uit het rapport niet concluderen of deze SO’s voortkomen uit ernstige bezwaren (major objections). De specifieke verplichtingen en aanbevelingen over de kwaliteit zijn allemaal vragen naar aanvullende gegevens die verder bevestigen dat de kwaliteit van het Pfizer-vaccin voldoet aan de gestelde eisen. De initiële gegevens gaven al aan dat de kwaliteit van het Pfizer-vaccin voldoet om het product voorwaardelijk te kunnen accepteren. Voor een definitieve handelsvergunning wordt verdere bevestiging noodzakelijk geacht.
Bent u van mening dat de voorwaardelijke handelsvergunning voor het Pfizer-vaccin moet worden ingetrokken indien niet (tijdig) aan deze specifieke verplichtingen en aanbevelingen wordt voldaan? Zo nee, waarom niet?
Dit is een wettelijke kwestie, geen kwestie van meningen. Als de fabrikant niet voldoet aan de gestelde voorwaarden, zal de Europese Commissie op advies van beoordelingscomités van het EMA (CHMP, PRAC) de (voorwaardelijke) handelsvergunning opschorten of andere maatregelen nemen. Dit werkt dan van rechtswege door in Nederland.
Op welke wijze zorgt u dat u op de hoogte blijft van de voortgang van de oplevering van de SO's en de opvolging van de aanbevelingen?
Hier word ik regelmatig over geïnformeerd door het College ter Beoordeling van Geneesmiddelen (hierna: CBG).
Kunt u inzicht geven in de voortgang van de oplevering van de SO's en van de opvolging van de aanbevelingen?
Een overzicht van de speciale verplichtingen (SO’s) en de voortgang van de oplevering voor het Pfizer-vaccin staat weergegeven in onderstaande tabel. De informatie over Specific Obligations is openbaar en is te vinden in Annex E van de SmPC en het EPAR, te vinden op de EMA-website.9
|
Module |
Description |
Due Date |
Progress |
|---|---|---|---|
|
Quality |
In order to complete the characterisation of the active substance and finished product, the MAH should provide additional data. |
July 2021. Interim- reports: 31 March 2021 |
Data door firma ingediend; beoordeling door EMA loopt. |
|
Quality |
In order to ensure consistent product quality, the MAH should provide additional information to enhance the control strategy, including the active substance and finished product specifications. |
July 2021. Interim- reports: 31 March 2021 |
Data door firma ingediend; beoordeling door EMA loopt. |
|
Quality |
In order to confirm the consistency of the finished product manufacturing process, the MAH should provide additional validation data. |
March 2021 |
Data door firma ingediend; voldaan. |
|
Quality |
In order to confirm the purity profile and ensure comprehensive quality control and batch-to-batch consistency throughout the lifecycle of the finished product, the MAH should provide additional information about the synthetic process and control strategy for the excipient ALC-0315. |
July 2021. Interim- reports: January 2021, April 2021. |
Data door firma ingediend; beoordeling door EMA loopt. |
|
Quality |
In order to confirm the purity profile and ensure comprehensive quality control and batch-to-batch consistency throughout the lifecycle of the finished product, the MAH should provide additional information about the synthetic process and control strategy for the excipient ALC-0159. |
July 2021. Interim- reports: January 2021, April 2021. |
Data door firma ingediend; beoordeling door EMA loopt. |
|
Clinical |
In order to confirm the efficacy and safety of Comirnaty, the MAH should submit the final Clinical Study Report for the randomized, placebo-controlled, observer-blind study C4591001. |
December 2023 |
Loopt nog, finale klinische onderzoeksrapport wordt december 2023 verwacht. |
De specifieke verplichtingen en aanbevelingen over de kwaliteit zijn allemaal vragen naar aanvullende gegevens die bevestigen dat de kwaliteit van het Pfizer-vaccin voldoet.
De gegevens gaven al aan dat de kwaliteit van het Pfizer-vaccin voldoet om het product voorwaardelijk te kunnen accepteren. Voor een definitieve handelsvergunning werd verdere bevestiging noodzakelijk geacht.
De firma heeft alle aanvullende gegevens betreffende de kwaliteit geleverd zoals gevraagd in de specifieke verplichtingen (op 2 augustus 2021). Een deel van de gegevens is al beoordeeld en voldoen aan de eisen. De procedures van beoordeling van een ander deel van de ingediende gegevens lopen nog.
Ook betreffende de aanbevelingen heeft de firma aan haar verplichtingen voldaan. Een aantal aanbevelingen loopt nog, omdat ze termijnen hebben later dan oktober 2021. Ditzelfde geldt voor de klinische specifieke verplichting: het opleveren van het finale klinische onderzoeksrapport voor studie C4951001.
Geen van de verstrekte aanvullende gegevens heeft tot een herziening van een inzicht geleid betreffende de kwaliteit en controle van het vaccin.
Weet u dat de veiligheidsbeoordeling van het Pfizer-vaccin in het EMA-rapport gebaseerd is op gegevens uit de eerste maanden van de nog lopende fase 2/3 studie, waarvan de langst lopende beschikbare follow-up tijd 12–13 weken na de tweede prik was?
Het klopt dat de initiële beoordeling van veiligheid gebaseerd is op een follow up tijd van zo’n drie maanden. De meeste bijwerkingen van vaccins treden op binnen 4–6 weken na vaccinatie. Zoals EMA ook heeft aangegeven in hun reflectie document «EMA considerations on COVID-19 vaccine approval, EMA/592928/2020» zou in principe een voorwaardelijke vergunning voor het in de handel brengen van een COVID-19-vaccin gebaseerd kunnen worden op beoordeling van veiligheidsgegevens van ten minste 6 weken na vaccinatie.10 Zoals gezegd was de follow-up tijd van de initiële veiligheidsbeoordeling van het Pfizer-vaccin tweemaal zo lang – de eerder genoemde ± drie maanden.
Inmiddels zijn de preregistratie gegevens aangevuld met post-registratie follow-up gegevens uit studies en spontane meldingen van de afgelopen 10 maanden. De post-registratie ervaring is gebaseerd op naar schatting 1.5 miljard toegediende doses van het Pfizer-vaccin.
Weet u dat in de bijlage van Richtlijn 75/318/EEG, deel III, hoofdstuk III staat dat «klinische verklaringen over de therapeutische werking en het niet schadelijke karakter bij normaal gebruik van een specialiteit niet kunnen worden geaccepteerd als adequaat bewijs als ze niet wetenschappelijk zijn gestaafd»?11
Het klopt dat deze passage in de bijlage staat, maar gegeven dat Richtlijn 75/318/EEG sinds 17 december 2001 niet meer in werking is, ontgaat mij de relevantie hiervan.
Bent u het ermee eens dat er momenteel nog onvoldoende wetenschappelijke basis is om een gedegen inschatting te kunnen maken van de veiligheid van het vaccin teneinde dit zonder risico toe te kunnen dienen aan het grootste deel van de Nederlandse bevolking? Zo nee, waarom niet?
Hier ben ik het niet mee eens. In Europa hebben we strenge wettelijke eisen rondom kwaliteit, werkzaamheid en veiligheid waaraan een potentieel geneesmiddel moet voldoen voordat het op de markt wordt toegelaten. Het Pfizer-vaccin voldoet aan deze eisen.
Weet u dat het EMA-rapport een Risico Management Plan bevat, waarin een opsomming wordt gegeven van de belangrijke veiligheidsrisico's en de nog ontbrekende informatie met betrekking tot de veiligheid van het vaccin en dat de indiener van de aanvraag, naast een aantal routinemaatregelen, geen aanvullende maatregelen genomen heeft om de geïdentificeerde risico's te minimaliseren?
Voor elk geneesmiddel of vaccin dat tot de markt wordt toegelaten op basis van een positieve baten-risico balans is de vergunninghouder wettelijk verplicht een Risico Management Plan (RMP) in te dienen als vast onderdeel van het registratiedossier.
Dit RMP geeft informatie over het veiligheidsprofiel van een geneesmiddel, beschrijft de activiteiten van de handelsvergunninghouder om tijdens de post- marketingperiode het veiligheidsprofiel verder te karakteriseren (activiteiten op het gebied van Geneesmiddelenbewaking) en licht de maatregelen toe die worden genomen om de risico's van het geneesmiddel bij patiënten te voorkomen of tot een minimum te beperken (risicominimalisatie-maatregelen).
Een RMP moet worden ingediend als onderdeel van het dossier van alle nieuwe aanvragen voor geneesmiddelen (inclusief generieke middelen en geneesmiddelen die alleen voor de nationale markt worden aangevraagd) en wordt door registratie-autoriteiten beoordeeld voordat een geneesmiddel kan worden toegelaten tot de markt. Vanzelfsprekend is een RMP ook voor alle goedgekeurde COVID-19 vaccins vastgesteld.12
Het is niet uitzonderlijk dat bij nieuwe geneesmiddelen, dus ook vaccins, sprake is van ontbrekende informatie en dat er onzekerheid bestaat over bijvoorbeeld een aantal veiligheidsaspecten. Dit kan bijvoorbeeld komen doordat het onethisch is om bepaalde groepen patiënten te includeren in de klinische studies – denk aan zwangere vrouwen of kinderen. Met preklinische (dier)studies wordt desalniettemin zo veel mogelijk informatie verzameld om de risico’s zo goed mogelijk te kunnen kwantificeren.
Daarna wordt een risico-inschatting gemaakt, waarin bepaald wordt hoe zwaar de onzekerheden wegen. Aan de hand daarvan wordt bekeken welke consequenties die risico’s hebben voor de toepassing van het geneesmiddel.
In het geval van ernstige verwachte risico’s wordt de aanvrager verplicht om, in aanvulling op de routinemaatregelen (zoals o.a. de contra-indicaties, waarschuwingen en voorzorgsmaatregelen zoals vermeld in de productinformatie) aanvullende risicominimaliserende maatregelen te treffen.
In het geval van het Pfizer-vaccin bleken de risico’s dusdanig laag te zijn dat de vergunninghouder geen verplichtingen zijn opgelegd om aanvullende risicominimaliserende maatregelen te treffen.
Voor meer informatie over verplichtingen voor handelsvergunninghouders zie: https://www.cbg-meb.nl/onderwerpen/hv-verantwoordelijkheden-na-verlening-handelsvergunning
Voor meer informatie o.a. over risicominimalisatie maatregelen zie: https://www.cbg-meb.nl/onderwerpen/hv-risk-management-plan
Heeft u, als eindverantwoordelijke voor de volksgezondheid en dus voor de veiligheid van de toediening van dit vaccin aan de Nederlandse bevolking, wel maatregelen genomen om de veiligheidsrisico's van het vaccin te minimaliseren? Zo ja, welke?
Er zijn verschillende manieren waarop de veiligheid van geneesmiddelen wordt gemonitord in Nederland en Europa. Allereerst verzamelt Lareb meldingen van mogelijke bijwerkingen in Nederland en worden opvallende gevallen gemeld bij het CBG, waarna deze indien nodig in Europees verband worden besproken en onderzocht. Zo wordt ervoor gezorgd dat niet alleen mogelijke bijwerkingen in Nederland in het oog worden gehouden, maar ook die in andere landen. Ook zijn fabrikanten verplicht om regelmatig overzichten over veiligheid naar het EMA op te sturen, zodat de veiligheid doorlopend kan worden gemonitord. Als laatste zijn fabrikanten verplicht om, zoals in eerdere vragen al aan de orde kwam, hun onderzoeken door te zetten. Hiermee kan bijvoorbeeld worden gemonitord hoe lang de bescherming van het vaccin aanhoudt, of kan de inzetbaarheid van het vaccin bij andere doelgroepen, zoals zwangere vrouwen, worden onderzocht.
Weet u dat nagenoeg alle in het Risico Management Plan van het EMA-rapport genoemde veiligheidsstudies doorlooptijden hebben tot en met 2023 en sommige zelfs tot in 2024?
Weet u dat op de website van het Rijksinstituut voor Volksgezondheid en Milieu (RIVM) over de veiligheid van het vaccin staat dat veiligheid bovenaan staat, en dat bij twijfel over de veiligheid het vaccin niet mag worden toegelaten in Nederland?
Op de website van het RIVM heb ik deze specifieke aangehaalde formulering niet gevonden. Op de website van de rijksoverheid staat het volgende: «Veiligheid van het coronavaccin is het belangrijkste. Hier gelden strenge eisen voor. Net als bij andere vaccins. Bij twijfel over de veiligheid van een vaccin wordt het niet toegelaten in Nederland».13
Kunt u uitleggen op welke andere wetenschappelijke bronnen u zich baseert, aangezien u kennelijk heeft geconcludeerd dat er geen twijfel kan zijn over de veiligheid van het Pfizer-vaccin, en waarom u uw mening niet gewoon baseert op de wetenschappelijke informatie van de hiervoor in de EU aangestelde controlerende autoriteit?
Deze vraag is gebaseerd op een onjuiste aanname. Er kan vervolgonderzoek aanbevolen worden voor een geneesmiddel, zonder dat dit betekent dat er twijfel is over de kwaliteit, veiligheid en werkzaamheid. Nederland volgt het oordeel van het EMA, waaraan het eerdergenoemde onderzoeksrapport aan ten grondslag ligt.
Weet u dat de in vraag 16 genoemde nog lopende fase 2/3 studie volgens het EMA-rapport in de «real world» wordt voortgezet en dat het eindrapport van deze studie moet worden opgeleverd op 31 augustus 2023?
In het kader van de afgegeven voorwaardelijke vergunning voor het Pfizer-vaccin, moet de vergunninghouder binnen het vastgestelde tijdschema o.a. voor december 2023 de werkzaamheid en veiligheid van het Pfizer-vaccin bevestigen middels de indiening van het laatste klinische onderzoeksrapport voor het gerandomiseerde, placebogecontroleerde, waarnemergeblindeerde onderzoek C4591001. Dit onderzoek betreft opvolging binnen de onderzoekspopulatie onder het lopende onderzoeksprotocol.
Zijn tot 31 augustus 2023 alle Nederlandse ontvangers van het Pfizer-vaccin bij de in vraag 25 genoemde studie betrokken?
De betreffende fase 2/3 studie (C4591001) heeft deelnemers tussen april en november 2020 ingesloten. Alleen deze deelnemers worden voor een periode van 2 jaar opgevolgd. Er waren geen Nederlandse centra betrokken bij de genoemde studie.
Indien het antwoord op vraag 26 «ja» is, op welke wijze wordt getoetst op de goede klinische praktijken en gecontroleerd op de wijze waarop gegevens, informatie en documenten worden verkregen, geregistreerd en gerapporteerd, welke volgens Richtlijn 2001/20/EG van 4 april 2001 strikt noodzakelijk zijn om de deelname van mensen aan de klinische proeven te rechtvaardigen?14
Ondanks dat het antwoord op vraag 26 «nee» is, wil ik hier het volgende aangeven. Een handelsvergunning voor geneesmiddelen wordt in de EU slechts verleend als het EMA of een nationale bevoegde autoriteit (in Nederland het CBG) heeft geoordeeld dat bij het in de vergunningsaanvraag opgenomen klinisch onderzoek heeft voldaan aan goede klinische praktijken als zijn vastgelegd in hoofdstuk 2 van Richtlijn nr. 2005/28/EG. Daarbij heeft een ethische commissie en/of de ter zake bevoegde autoriteit voorafgaand aan een klinische proef het bijbehorende onderzoeksprotocol getoetst op het voldoen aan goede klinische praktijken. Verder houden daartoe bevoegde autoriteiten toezicht op de naleving van de regels voor klinisch onderzoek. Een van die regels is bijvoorbeeld dat het gebruik van persoonsgebonden (vaccinatie)gegevens voor onderzoeksdoeleinden (bijvoorbeeld centrale registratie en gegevensuitwisseling met RIVM (CIMS)) niet mogelijk is zonder expliciete toestemming (informed consent) van de patiënt/gebruiker of in dit geval, de gevaccineerde.
Indien het antwoord op vraag 26 «nee» is: aangezien de resultaten van in vraag 25 genoemde veiligheidsstudie pas op 31 augustus 2023 en de overige veiligheidsstudies ook pas in 2023 en 2024 worden opgeleverd, op welke grond en onder welke voorwaarden wordt het middel tot die tijd op experimentele basis toegediend aan Nederlanders die niet bij de studies betrokken zijn?
De stelling dat het vaccin op experimentele basis wordt toegediend is onjuist. Het Pfizer-vaccin is door de bevoegde autoriteit veilig en werkzaam bevonden en daarnaast zijn er ook aanbevelingen voor vervolgonderzoek gegeven. Dit is geen tegenstelling. De meeste bijwerkingen van vaccins treden op binnen 4–6 weken na vaccinatie. Zoals het EMA ook heeft aangegeven in het reflectiedocument «EMA considerations on COVID-19 vaccine approval, EMA/592928/2020» zou in principe een voorwaardelijke vergunning voor het in de handel brengen van een COVID-19-vaccin gebaseerd kunnen worden op beoordeling van veiligheidsgegevens van ten minste 6 weken na vaccinatie. De follow-up tijd van de initiële veiligheidsbeoordeling was tweemaal zo lang – ongeveer drie maanden.
Heeft u, als eindverantwoordelijke voor de volksgezondheid en dus voor de veiligheid van de toediening van deze vaccins aan de Nederlandse bevolking, maatregelen genomen voor het toetsen en controleren van de werking en veiligheid van de vaccins die op experimentele basis worden toegediend aan de Nederlandse bevolking? Zo nee, waarom niet?
Allereerst benoem ik graag opnieuw dat de stelling dat het vaccin op experimentele basis wordt toegediend pertinent onjuist is.
Dat gezegd hebbende, zijn er verschillende manieren waarop de veiligheid van geneesmiddelen wordt gemonitord in Nederland en Europa, hiervoor verwijs ik naar de hierboven gegeven antwoorden.
Zie hiervoor mijn antwoord op vraag 20, waarin ik reeds een overzicht heb gegeven van genomen maatregelen.
Weet u dat het gebruik van het Pfizer-vaccin bij zwangerschap en borstvoeding in het Risico Management Plan in het EMA-rapport onder het kopje «ontbrekende informatie» wordt genoemd en dat de uit te voeren studies om de veiligheid op dit punt te onderzoeken moeten worden opgeleverd in 2023 en 2024?
Er is beperkte ervaring met het gebruik van het Pfizer-vaccin bij zwangere vrouwen aangezien deze bij de pre-registratie (fase 2/3) studie waren uitgesloten van deelname (zoals gebruikelijk is bij veel geneesmiddelen). De fabrikant is verplicht om hier aanvullende informatie over te verzamelen in post-marketing studies en maandelijkse veiligheidsrapportages. Uit dierproeven zijn er geen aanwijzingen dat het vaccineren van zwangeren invloed heeft op de zwangerschap of het ongeboren kind. Ook geeft de beoordeling van post-marketing gevallen van blootstelling aan het Pfizer-vaccin tijdens de zwangerschap of borstvoeding geen aanleiding tot ongerustheid. Er zijn geen aanwijzingen voor nadelige effecten op de zwangerschap of het pasgeboren kind gevonden. Gezien het belang van het bevestigen van de veiligheid van gebruik van de COVID-19 vaccins gedurende de zwangerschap geeft het EMA ondersteuning aan projecten met als doel het volgen van de veiligheid van COVID-19 vaccins gedurende de zwangerschap, bijvoorbeeld door het ConcePTION consortium (www.imi-conception.eu).
Weet u dat op de website van het RIVM staat dat zwangere vrouwen het advies krijgen om zich te laten vaccineren tegen het coronavirus met de mRNA-vaccins van Pfizer en Moderna, en dat vaccinatie veilig en effectief zou zijn, ook in de zwangerschap?
Kunt u uitleggen op welke andere wetenschappelijke bron het advies van de overheid voor zwangere vrouwen is gebaseerd, en waarom het advies niet gewoon wordt gebaseerd op de wetenschappelijke informatie van de hiervoor in de EU aangestelde controlerende autoriteit?
Het advies rondom vaccineren tijdens de zwangerschap is opgesteld door een multidisciplinaire werkgroep met afgevaardigden vanuit de Nederlandse Vereniging voor Obstetrie & Gynaecologie (NVOG), Koninklijke Nederlandse Organisatie van Verloskundigen (KNOV), Nederlandse Vereniging voor Kindergeneeskunde (NVK), Nederlandse Vereniging voor Medische Microbiologie (NVMM), Patiëntenfederatie Nederland, het Rijksinstituut voor Volksgezondheid en Milieu (RIVM) en de Vereniging voor Hygiëne & Infectiepreventie in de Gezondheidszorg (VHIG).15 Bij het opstellen van dit advies hebben deze partijen zich gebaseerd op de geldende richtlijnen en de laatste stand van de wetenschap.
Kunt u aangeven welke maatregelen u heeft genomen om de veiligheid te waarborgen en monitoren van de toepassing van het Pfizer-vaccin op mensen met een zwakke gezondheid en onderliggend lijden, aangezien in het EMA-rapport staat dat het niet is getest op deze groep en deze groep in het Risico Management Plan valt onder het kopje «ontbrekende informatie»?
In het EMA-rapport is aangegeven dat in beide onderzoeksgroepen (dat wil zeggen: de vaccin-groep en placebogroep) 20.5% van de deelnemers «comorbiditeiten» (onderliggende aandoeningen) hadden. Dit waren bijvoorbeeld mensen met diabetes, obesitas of longaandoeningen. Zoals ik in eerdere antwoorden, waaronder het antwoord op vraag 30, aangeef, zijn aanbevelingen voor vervolgonderzoek standaard.
Weet u dat de onderzoekers na het uitvoeren van de effectiviteitsstudies geconcludeerd hebben dat een wetenschappelijke onderbouwing ontbreekt om een exacte inschatting te kunnen maken of het Pfizer-vaccin ernstige ziekte kan voorkomen, dat ernstige ziekte zelden voorkomt en dat om die reden de effectiviteit van het Pfizer-vaccin op dat punt niet met voldoende statistische bewijslast kan worden aangetoond?
Ten tijde van de afgifte van de voorwaardelijke handelsvergunning van het vaccin waren er inderdaad te weinig cases van ernstige ziekte om een betrouwbare effectschatting vast te stellen voor het voorkomen van ernstige ziekte door COVID-19. Wel kwam uit de studieresultaten naar voren dat er minder cases in de gevaccineerde groep waren dan in de placebogroep. Omdat de studie nog verder doorliep was het de verwachting dat er op korte termijn wel voldoende gegevens beschikbaar zouden komen om de mate van bescherming tegen ernstige ziekte vast te kunnen stellen. Deze informatie is inmiddels inderdaad aangeleverd – in mijn antwoord op vraag 36 ga ik hier in meer detail op in.
Weet u dat het EMA-rapport stelt dat ernstige ziektegevallen in de studie nagenoeg niet voorkwamen, en dat bewijs voor een beschermend effect van het Pfizer-vaccin tegen ernstige ziekte derhalve ontbreekt?
Voor alle COVID-vaccins die tot nu toe een goedkeuring hebben gekregen, is werkzaamheid vastgesteld op basis van bescherming tegen symptomatische infectie in grote gerandomiseerde fase 3 studies. Voordat de studies gestart zijn, is met de verschillende regulators (o.a. EMA, FDA, MHRA etc.) afgesproken dat het aantonen van bescherming tegen symptomatische infectie acceptabel is. Bij alle deelnemers die na vaccinatie (met het actieve vaccin of met een placebo) COVID-achtige klachten kregen, is een PCR test afgenomen om te kijken of ze besmet waren met het SARS-CoV-2 virus. Hieruit bleek dat de vaccins bescherming bieden tegen symptomatische COVID-19. Een klein deel van deze mensen kreeg ernstige klachten, te weinig om hier een specifiek werkzaamheidspercentage voor vast te stellen. Om specifiek werkzaamheid tegen ernstige ziekte aan te tonen, hadden de studies nog veel groter moeten zijn en/of zou het langer duren voordat er voldoende cases optraden om een betrouwbaar werkzaamheidspercentage vast te stellen.
Ten tijde van de (voorwaardelijke) goedkeuring van het vaccin was geen effectschatting mogelijk op het voorkomen van ernstige ziekte door COVID-19. Omdat de studie nog verder doorliep was het de verwachting dat er op korte termijn wel voldoende gegevens over de mate van bescherming tegen ernstige ziekte beschikbaar zouden komen. Deze informatie is kort na de voorwaardelijke goedkeuring aangeleverd en direct beoordeeld.
De geüpdatete data uit de lopende fase 2/3 studie die na goedkeuring beschikbaar zijn gekomen laten hoge werkzaamheid zien tegen ernstig beloop van COVID-19. Dit is opgenomen in rubriek 5.1 van de SmPC.16
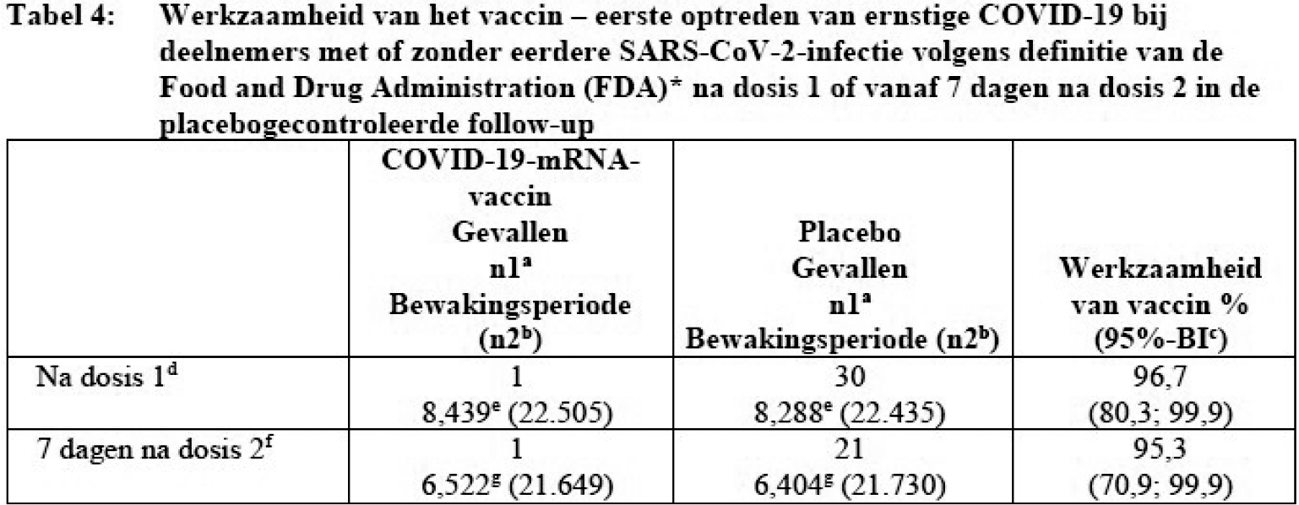
Opmerking: Bevestigde gevallen werden bepaald met behulp van Reverse Transcription-Polymerase Chain Reaction (RT-PCR); ten minste 1 symptoom stemde overeen met COVID-19 (symptomen waren: koorts; nieuwe of toegenomen hoest; nieuwe of toegenomen kortademigheid; koude rillingen; nieuwe of toegenomen spierpijn; nieuw verlies van smaakzin of reukzin; keelpijn; diarree; braken).
Ernstige ziekte van COVID-19 zoals gedefinieerd door de FDA is bevestigde COVID-19 en aanwezigheid van ten minste 1 van de volgende:
– klinische tekenen in rusttoestand die duiden op een ernstige systemische ziekte (ademhalingsritme ≥ 30 ademhalingen per minuut, hartslag ≥ 125 slagen per minuut, zuurstofsaturatie ≤ 93% bij kamerlucht op zeeniveau, of verhouding van partiële druk van arteriële zuurstof ten opzichte van fractionele geïnspireerde zuurstof < 300 mmHg);
– ademhalingsfalen (gedefinieerd als noodzaak van high-flow zuurstof, niet-invasieve beademing, kunstmatige beademing of extracorporele membraanoxygenatie [ECMO]);
– bewijs van shock (systolische bloeddruk < 90 mmHg, diastolische bloeddruk < 60 mmHg of noodzaak van vasopressoren);
– significante acute nier-, lever- of neurologische disfunctie;
– opname op een intensive-careafdeling;
– overlijden.
Op grond van welke informatie hebben het RIVM en u vanaf het begin van ingebruikneming in Nederland gezegd dat het Pfizer-vaccin vooral goed beschermt tegen ernstige ziekte? Kunt u uitleggen waarom de informatie die u hierover verstrekt aan de Nederlandse burgers niet gebaseerd is op de wetenschappelijke studies die de daarvoor aangestelde controlerende EU-autoriteit hanteert, maar op meningen en aannames die deze wetenschappelijke studies tegenspreken?
Weet u dat in de bijlage van richtlijn 91/507/EEG, deel 4 onder F staat dat het betrekken van een groot aantal proefpersonen bij een onderzoek niet mag worden beschouwd als een adequate vervanging voor een onderzoek met een deugdelijke controleopzet, en dat voor de handelsvergunning de resultaten van deugdelijk wetenschappelijk onderzoek dus moeten prevaleren boven een inschatting van de werking in een zeer groot deel van de bevolking?17
Het klopt dat deze tekst in de bijlage bij deze richtlijn staat, maar gegeven dat voor Richtlijn 91/507/EEG geldt dat deze sinds 17 december 2001 niet meer in werking is, ontgaat mij de relevantie hiervan.
Kunt u uitleggen waarom u het coronavirus nog steeds onder de Wet publieke gezondheid vindt vallen als een A-statusziekte, terwijl in het EMA-rapport door de wetenschappelijke studies is bevestigd dat ernstige ziekte door het coronavirus zeer zeldzaam is?
Zoals eerder aangegeven in de beantwoording van vragen die zijn gesteld op 21 september 202018 is de indeling van een infectieziekte in groep A, in de Wet publieke gezondheid (hierna: Wpg) gerelateerd aan het volgende:
– De vraag of landelijke regie door de Minister van Volksgezondheid, Welzijn en Sport (hierna: VWS) noodzakelijk wordt geacht. Als dat het geval is, dan wordt de ziekte in groep A ingedeeld.
– De maatregelen gericht op individuen die potentieel noodzakelijk geacht worden om te kunnen worden opgelegd ter bescherming van de publieke gezondheid. Voor de B2-ziekten is dat een tijdelijk beroepsverbod. Voor B1 ziekten kan daarnaast een gedwongen onderzoek en gedwongen isolatie worden opgelegd en voor A-ziekten komt daar nog de mogelijkheid voor het opleggen van een gedwongen quarantaine bij. De C-ziekten kennen geen op het individu gerichte gedwongen maatregelen, maar alleen advisering en begeleiding van individuen en groepen.
– Voor alle categorieën geldt een meldingsplicht, waarbij voor een A-ziekte de melding onverwijld wordt doorgegeven aan de Gemeentelijke gezondheidsdienst (hierna: GGD).
Door het Rijksinstituut voor Volksgezondheid en Milieu (hierna: RIVM) is een beslisschema opgesteld met een aantal vragen om te bepalen of een meldingsplicht geldt voor een infectieziekte. Hiervan is sprake als, kort weergeven:
– Het gaat om internationale afspraken, bijvoorbeeld dat de infectieziekte mogelijk internationale consequenties heeft en moet worden gemeld aan de WHO volgens de Internationale Gezondheidsregeling.
– De effectiviteit, bijvoorbeeld of er een kans is op substantiële morbiditeit en/of een potentieel capaciteitsprobleem voor ziekenhuizen en huisartsen, dan wel de vraag of de infectieziekte voortkomt uit een open bron waartegen de omgeving zich niet of moeilijk kan beschermen.
– De haalbaarheid, bijvoorbeeld of de werklast voor de publieke gezondheidszorgdiensten proportioneel is aan de uitkomsten voor de publieke gezondheid.
– Tot slot de noodzaak, bijvoorbeeld of de meldingsplicht onmisbaar is voor preventie en bestrijding en niet op een andere manier informatie kan worden verkregen.
Het volledige beslisschema bestaat sinds 2015 en is te raadplegen via de volgende link: https://magazines.rivm.nl/2019/01/infectieziekten-bulletin/de-meldingsplichtprocedure-van-aanvraag-tot-wet. De door u aangehaalde reden is derhalve niet doorslaggevend voor de indeling van een infectieziekte in Wpg systematiek.
Kunt u uitleggen waarom u nog steeds de bevolking blijft waarschuwen voor het risico op ernstige ziekte door het coronavirus, terwijl in het EMA-rapport door de wetenschappelijke studies is bevestigd dat ernstige ziekte door het coronavirus zeer zeldzaam is?
Zoals aangegeven in de brief van 22 november 2021 is in de afgelopen periode het epidemiologisch beeld verslechterd. Het reproductiegetal, de snelheid waarmee het virus zich verspreidt, is nu 1,26 (95%-interval 1.24 – 1.28) (ijkdatum: 4 november). In de afgelopen 7 kalenderdagen (11-18 november) is het aantal meldingen van SARS-CoV-2-positieve personen gestegen met 46% in vergelijking met de 7 dagen ervoor. In de afgelopen 7 kalenderdagen werden landelijk 716 personen per 100.000 inwoners gemeld met een positieve test voor SARS-CoV-2, vergeleken met 492 per 100.000 inwoners in de week daarvoor, een stijging van 46%. De instroom en bezetting op verpleegafdelingen en IC in de ziekenhuizen van personen met een positieve test op SARS-CoV-2 nam toe. In de afgelopen kalenderweek werden 1476 opnames in het ziekenhuis geregistreerd, waarvan 228 op de IC, vergeleken met respectievelijk 1252 en 209 de week daarvoor (bron: stichting NICE, dd 18 november). Dit is een stijging van 18% instroom totaal, en van 9% op de IC. De hoogste aantallen opgenomen COVID-10 patiënten op verpleegafdelingen in het ziekenhuis betreffen personen van boven de 80 jaar, op de IC van 60–79 jaar. Op 18 november 2021 was de bedbezetting 2110: 1697 op de verpleegafdelingen in het ziekenhuis en 413 op de IC (bron: LCPS). De impact van het virus op de samenleving is derhalve groot en het aantal IC- en ziekenhuisopnamen geeft aan dat de ziekte voor bepaalde personen een ernstig ziekteverloop kent.
Weet u dat in het EMA-rapport wordt vermeld dat er tijdens de beoordeling diverse problemen werden geconstateerd met betrekking tot de kwaliteitsstatus van de drie productie- en testlocaties van het Pfizer-vaccin, en dat deze problemen werden aangemerkt als ernstige bezwaren, maar dat de locaties toch het benodigde kwaliteitscertificaat kregen en dat daarbij wordt verwezen naar het feit dat het Pfizer-vaccin vanwege de noodsituatie een tijdelijke uitzonderingsstatus heeft, waardoor de controle van het Pfizer-vaccin buiten de EU mag plaatsvinden?
Ja. Ten tijde van de voorwaardelijke toelating waren er nog geen testlocaties met een Good Manucturing Practices (GMP) certificaat beschikbaar binnen de EU. Hierover zijn ernstige bezwaren geuit. Na beantwoording door de firma mocht controle per uitzondering ook buiten de EU plaatsvinden en waren de ernstige bezwaren weggenomen. De vrijgiftetesten werden uitgevoerd in testlocaties in de VS die gecertificeerd zijn door de FDA, waarmee de EU een wederzijdse erkenningsovereenkomst heeft. In de maanden daarna zijn er gecertificeerde testlocaties in de EU bijgekomen (zie ook vraag19 en zijn de vrijgiftetesten in de EU uitgevoerd.
Weet u dat de in vraag 41 genoemde uitzonderingsstatus verviel op 31 augustus 2021 en dat vanaf die datum de controle plaats moet vinden binnen de EU?
Kunt u bevestigen dat de controle van de vaccins die in Nederland worden toegediend sinds 31 augustus 2021 binnen de EU plaatsvindt? Kunt u in dat geval aangeven waar dit plaatsvindt?
Ja, dat kan ik bevestigen. De controle van de vaccins die in Nederland worden toegediend, vindt sinds 31 augustus 2021 plaats binnen de EU. Na registratie van het Pfizer-vaccin, en voor 31 augustus 2021, zijn meerdere fabrikanten toegevoegd aan het registratiedossier, waaronder fabrikanten die verantwoordelijk zijn voor de controle van de vaccins; deze controlelaboratoria zijn gevestigd in Duitsland, Frankrijk, België, Ierland, Italië en Kroatië.
Bent u van mening dat de voorwaardelijke handelsvergunning voor het Pfizer-vaccin moet worden ingetrokken indien de controle van het Pfizer-vaccin nog steeds buiten de EU plaatsvindt? Zo nee, waarom niet?
Ik verwijs u naar mijn antwoord op de vorige vraag, waarin ik bevestig dat de controle plaatsvindt binnen de EU.
Op welke wijze ziet u erop toe dat de handelsvergunning in Nederland wordt geschorst of ingetrokken indien bij de jaarlijkse controle blijkt dat de indiener niet heeft voldaan aan tijdige oplevering van de voorwaarden?
Als de fabrikant niet voldoet aan de gestelde voorwaarden, zal de Europese Commissie op advies van beoordelingscomités van het EMA (CHMP, PRAC) de (voorwaardelijke) handelsvergunning opschorten of andere maatregelen nemen. Dit werkt dan van rechtswege door in Nederland.
Weet u dat in artikel 9 van Richtlijn 65/65/EEG staat: «De vergunning laat onverlet de uit het gemene recht voortvloeiende aansprakelijkheid van de fabrikant of, in voorkomend geval, van degene die voor het in de handel brengen van het product verantwoordelijk is»?20
Dit klopt, maar voor Richtlijn 65/65/EEG geldt dat deze sinds 17 december 2001 niet meer in werking is en daarom niet meer relevant is voor de huidige situatie.
Weet u dat in artikel 36 van Verordening 2309/93 staat dat door verlening van een vergunning geen afbreuk wordt gedaan aan de algemene burgerlijke en strafrechtelijke aansprakelijkheid in de Lidstaten van de fabrikant of, indien van toepassing, van de voor het in de handel brengen van het geneesmiddel verantwoordelijke persoon?21
Zoals ik in het antwoord op uw vierde vraag heb aangegeven, is verordening (EEG) 2309/93 sinds 19 november 2005 niet meer in werking. Het klopt wel dat deze passage in de vervallen verordening stond.
Kunt u bevestigen dat de in vraag 46 en 47 genoemde artikelen onverkort van toepassing zijn op de voorwaardelijke handelsvergunning op grond waarvan de COVID-19-vaccins in Nederland worden toegediend aan burgers? Zo nee, waarom niet?
Zoals ik in de vorige twee vragen aangaf, zijn deze richtlijn en verordening niet meer in werking en daarom niet van toepassing op enigerlei situatie die na 17 december 2001 respectievelijk 19 november 2005 heeft plaatsgevonden.
Wie is de in vraag 46 en 47 genoemde persoon die verantwoordelijk is voor het in Nederland in de handel brengen van de COVID-vaccins?
Wie draagt de in vraag 46 en 47 genoemde aansprakelijkheid voor het gebruik van de COVID-vaccins in Nederland?
Wilt u deze vragen afzonderlijk van elkaar beantwoorden, vóór aanvang van het eerstvolgende debat over de ontwikkelingen rondom het coronavirus?
Ik heb de vragen zo veel als mogelijk afzonderlijk beantwoord, maar het is helaas niet gelukt om aan het tweede deel van uw verzoek te voldoen.
EMA, 19 februari 2021, «Assessment report – COVID-19 mRNA vaccine (nucleoside-modified)» (www.ema.europa.eu/en/documents/assessment-report/comirnaty-epar-public-assessment-report_en.pdf).
Bureau voor publicaties van de Europese Unie, 26 januari 1965, «Richtlijn 65/65/EEG van de Raad van 26 januari 1965 betreffende de aanpassing van de wettelijke en bestuursrechtelijke bepalingen inzake farmaceutische specialiteiten» (op.Europa.eu/nl/publication-detail/-/publication/a761f2b9-d398-4fc5-a2fe-f1f7bebdb9b4/).
Bureau voor publicaties van de Europese Unie, 20 mei 1975, «Richtlijn 75/318/EEG van de Raad van 20 mei 1975 betreffende de onderlinge aanpassing van de wetgevingen van de Lid-Staten inzake de analytische, toxicologisch- farmacologische en klinische normen en voorschriften betreffende proeven op farmaceutische specialiteiten» (op.Europa.eu/en/publication-detail/-/publication/845d1ac1-0b12-4d7f-8cbc-821ae51a6b2d/language-nl).
Publikatieblad van de Europese Gemeenschappen, 9 juni 1975, «Tweede richtlijn van de Raad van 20 mei 1975 betreffende de aanpassing van de wettelijke en bestuursrechtelijke bepalingen inzake farmaceutische specialiteiten (75/319/EEG)» (https://eur-lex.europa.eu/legal-content/NL/TXT/PDF/?uri=CELEX:31975L0319&from=nl).
Publikatieblad van de Europese Gemeenschappen, 26 september 1991, «Richtlijn van de Commissie van 19 juli 1991 tot wijziging van de bijlage van Richtlijn 75/318/EEG van de Raad betreffende de onderlinge aanpassing van de wetgevingen van de Lid-Staten inzake de analytische, toxicologisch-farmacologische en klinische normen en voorschriften betreffende proeven op farmaceutische specialiteiten (91 /507/EEG)» (https://eur-lex.europa.eu/legal-content/NL/TXT/PDF/?uri=CELEX:31991L0507&from=NL).
Publikatieblad van de Europese Gemeenschappen, 24 augustus 1993, «Verordening (EEG) Nr. 2309/93 van de Raad van 22 juli 1993 tot vaststelling van communautaire procedures voor het verlenen van vergunningen voor en het toezicht op geneesmiddelen voor menselijk en diergeneeskundig gebruik en tot oprichting van een Europees Bureau voor de geneesmiddelenbeoordeling» (https://eur-lex.europa.eu/legal-content/NL/TXT/PDF/?uri=CELEX:31993R2309&from=NL).
Art. 14 -bis van Verordening (EG) Nr. 726/2004 tot vaststelling van procedures van de Unie voor het verlenen van vergunningen en het toezicht op geneesmiddelen voor menselijk en diergeneeskundig gebruik en tot oprichting van een Europees Geneesmiddelenbureau.
Publikatieblad van de Europese Gemeenschappen, 26 september 1991, «Richtlijn van de Commissie van 19 juli 1991 tot wijziging van de bijlage van Richtlijn 75/318/EEG van de Raad betreffende de onderlinge aanpassing van de wetgevingen van de Lid-Staten inzake de analytische, toxicologisch-farmacologische en klinische normen en voorschriften betreffende proeven op farmaceutische specialiteiten (91 /507/EEG)» (https://eur-lex.europa.eu/legal-content/NL/TXT/PDF/?uri=CELEX:31991L0507&from=NL).
Te vinden op: https://www.ema.europa.eu/en/documents/product-information/comirnaty-epar-product-information_en.pdf
Te vinden op: https://www.ema.europa.eu/en/documents/other/ema-considerations-covid-19-vaccine-approval_en.pdf
Bureau voor publicaties van de Europese Unie, 20 mei 1975, «Richtlijn 75/318/EEG van de Raad van 20 mei 1975 betreffende de onderlinge aanpassing van de wetgevingen van de Lid-Staten inzake de analytische, toxicologisch- farmacologische en klinische normen en voorschriften betreffende proeven op farmaceutische specialiteiten» (op.Europa.eu/en/publication-detail/-/publication/845d1ac1-0b12-4d7f-8cbc-821ae51a6b2d/language-nl).
Voor het Pfizer-vaccin te vinden op: Comirnaty, INN-COVID-19 mRNA Vaccine (nucleoside-modified) (europa.eu)
Publikatieblad van de Europese Gemeenschappen, 1 mei 2001 «Richtlijn 2001/20/EG van het Europees parlement en de Raad van 4 april 2001 betreffende de onderlinge aanpassing van de wettelijke en bestuursrechtelijke bepalingen van de lidstaten inzake de toepassing van goede klinische praktijken bij de uitvoering van klinische proeven metgeneesmiddelen voor menselijk gebruik» (https://eur-lex.europa.eu/legal-content/NL/TXT/PDF/?uri=CELEX:32001L0020).
https://www.nvog.nl/wp-content/uploads/2021/07/Standpunt-NVOG-Vaccinatie-COVID-19-zwangerschap.pdf
Te vinden op: https://www.ema.europa.eu/en/documents/product-information/comirnaty-epar-product-information_en.pdf
Publikatieblad van de Europese Gemeenschappen, 26 september 1991, «Richtlijn van de Commissie van 19 juli 1991 tot wijziging van de bijlage van Richtlijn 75/318/EEG van de Raad betreffende de onderlinge aanpassing van de wetgevingen van de Lid-Staten inzake de analytische, toxicologisch-farmacologische en klinische normen en voorschriften betreffende proeven op farmaceutische specialiteiten (91 /507/EEG)» (https://eur-lex.europa.eu/legal-content/NL/TXT/PDF/?uri=CELEX:31991L0507&from=NL).
Publikatieblad van de Europese Gemeenschappen, 26 september 1991, «Richtlijn van de Commissie van 19 juli 1991 tot wijziging van de bijlage van Richtlijn 75/318/EEG van de Raad betreffende de onderlinge aanpassing van de wetgevingen van de Lid-Staten inzake de analytische, toxicologisch-farmacologische en klinische normen en voorschriften betreffende proeven op farmaceutische specialiteiten (91 /507/EEG)» (https://eur-lex.europa.eu/legal-content/NL/TXT/PDF/?uri=CELEX:31991L0507&from=NL).
Bureau voor publicaties van de Europese Unie, 26 januari 1965, «Richtlijn 65/65/EEG van de Raad van 26 januari 1965 betreffende de aanpassing van de wettelijke en bestuursrechtelijke bepalingen inzake farmaceutische specialiteiten» (op.Europa.eu/nl/publication-detail/-/publication/a761f2b9-d398-4fc5-a2fe-f1f7bebdb9b4/).
Publikatieblad van de Europese Gemeenschappen, 24 augustus 1993, «Verordening (EEG) Nr. 2309/93 van de Raad van 22 juli 1993 tot vaststelling van communautaire procedures voor het verlenen van vergunningen voor en het toezicht op geneesmiddelen voor menselijk en diergeneeskundig gebruik en tot oprichting van een Europees Bureau voor de geneesmiddelenbeoordeling» (https://eur-lex.europa.eu/legal-content/NL/TXT/PDF/?uri=CELEX:31993R2309&from=NL).
Kopieer de link naar uw clipboard
https://zoek.officielebekendmakingen.nl/ah-tk-20212022-923.html
De hier aangeboden pdf-bestanden van het Staatsblad, Staatscourant, Tractatenblad, provinciaal blad, gemeenteblad, waterschapsblad en blad gemeenschappelijke regeling vormen de formele bekendmakingen in de zin van de Bekendmakingswet en de Rijkswet goedkeuring en bekendmaking verdragen voor zover ze na 1 juli 2009 zijn uitgegeven. Voor pdf-publicaties van vóór deze datum geldt dat alleen de in papieren vorm uitgegeven bladen formele status hebben; de hier aangeboden elektronische versies daarvan worden bij wijze van service aangeboden.